Figures obtained from the manuscript with author’s consent.
By Chris Matheson (Group Leader, Chemistry)
Co-deletion of genes neighbouring a tumour suppressor gene locus can often be exploited therapeutically via synthetic lethality. Tango Therapeutics recently reported the discovery of TNG9081, an inhibitor taking advantage of one such co-deletion; that of methylthioadenosine phosphorylase (MTAP), resultant from the deletion of CDKN2A. The protein MTAP is involved in the methionine salvage pathway, recycling methionine from methylthioadenosine (MTA) – hence, MTAP deletion results in accumulation of MTA, which is an S-adenosyl-L-methionine (SAM)-competitive inhibitor of the type II arginine methyltransferase PRMT5. Given the role of PRMT5 in various cellular functions, including DNA-damage response (DDR), cell cycle progression and apoptosis, partial inhibition of PRMT5 via MTA accumulation sensitises MTAP-null cancer cells to further PRMT5 targeted inhibition.
Early PRMT5 inhibitors lacked selectivity for MTAP-null cancer cell lines due to their action via SAM-competitive or SAM-cooperative binding – SAM being the universal methyl donor for methyltransferases in healthy tissues. To avoid inhibiting PRMT5 in both MTAP-null and healthy tissues, Tango Therapeutics set out to identify MTAP-null selective inhibitors that bind PRMT-5 in an MTA-cooperative manner. Furthermore, due the frequency of MTAP-deletion in CNS tumours, including glioblastoma multiforme (GBM), it was considered desirable to obtain chemical matter that was suitable for development into blood-brain-barrier (BBB) penetrant therapies. To this end, a high-throughput screening (HTS) effort was performed against PRMT5 in the presence of both SAM and MTA. The authors describe the development of a hit series, from the HTS oxamide hit compound 1 (Figure 1).
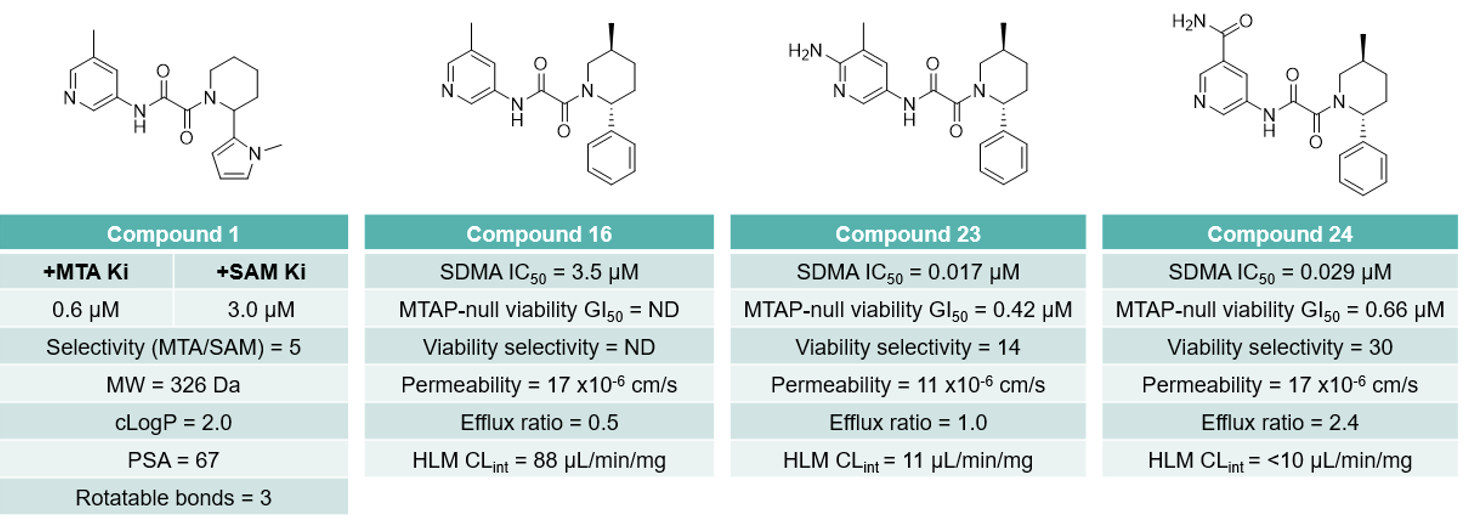
During the early phase of development, a co-crystal structure of the preferred R-enantiomer of compound 1 (1-R) bound to PRMT5 in the presence of MTA was solved, proving crucial in the design of selective analogues (Figure 2). Most importantly, it was observed that the pyridine methyl of 1-R locks Glu435 in the orientation preferred for MTA-binding, as opposed to the rotamer required to bind SAM. To further build on this, substitution on the pyridine was explored to improve interactions with the backbone carbonyl of Glu435 and drive selectivity for the PRMT5-MTA complex. Gratifyingly, the 6-amino (23) and 5-carboxamido (24) pyridines increased both cellular potency and selectivity over MTAP-WT cells (Figure 1).
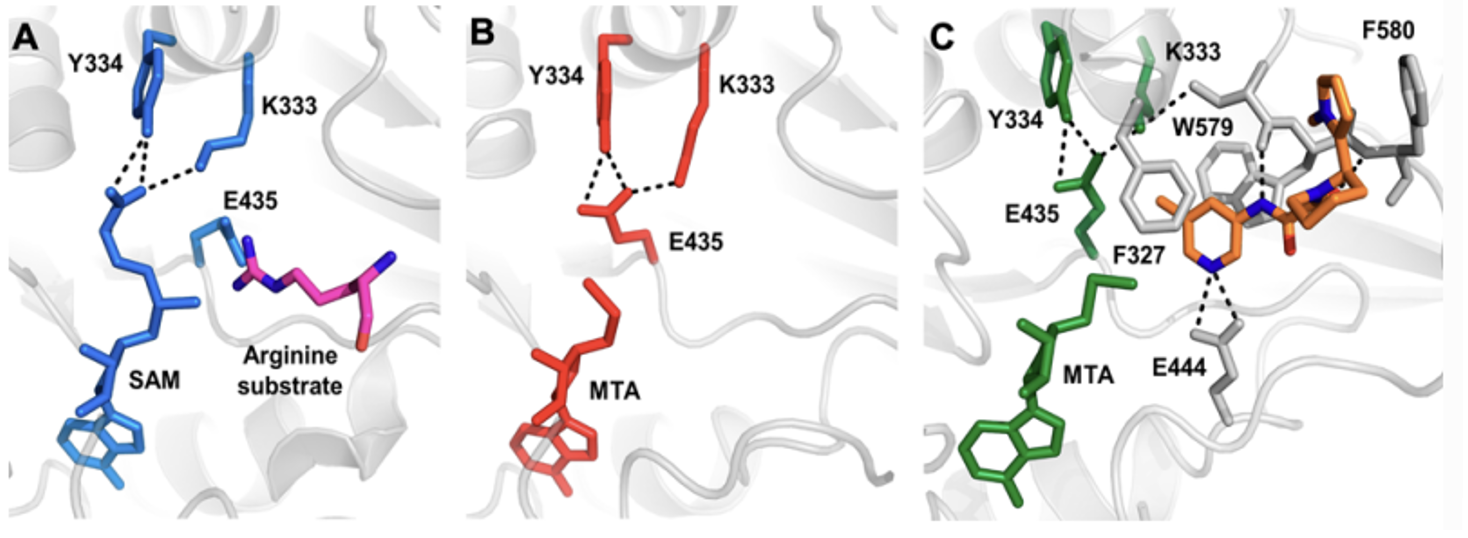
Structural biology assessment of both 23 and 24 revealed that the substitution around the piperidine adopted an unusual bis-axial conformation and that the phenyl ring in both cases was rotated relative to the pyrrole of compound 1, resulting in the phenyl being presented towards a backbone serine (Figure 3). As a proof-of-principle the relevant phenols were prepared, yielding improved viability, potency and increased selectivity for MTAP-null cells (data not shown). However, as anticipated, permeability and efflux suffered. An inspired solution to this problem was the introduction of a benzothiazole; a less commonly utilised phenol bioisostere, that was able to interact with the serine backbone carbonyl through the C-S σ* orbital, avoiding the introduction of an additional H-bond donor.
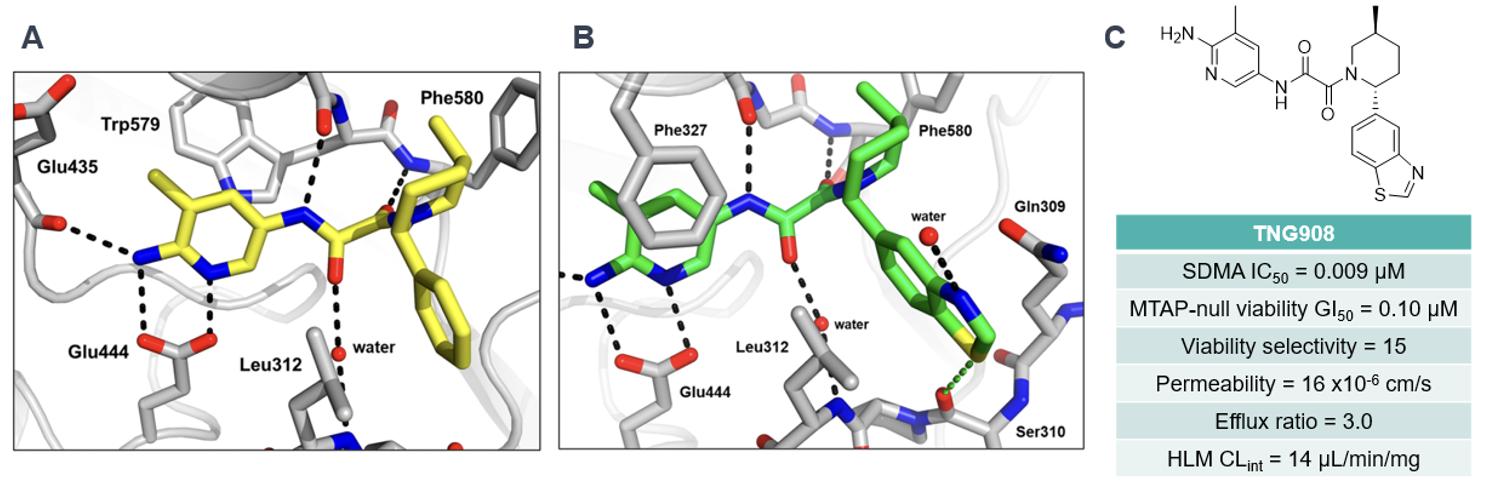
This culminated in TNG908, a compound with excellent potency and MTAP-null selectivity, with good permeability and stability in human liver microsomes (HLM). Furthermore, TNG908 showed good PK performance and was found to be BBB-penetrant in cynomolgus monkeys, prompting its nomination as a candidate in a Phase I/II clinical study against MTAP-null solid tumours (NCT05275478). The significant gains in potency and selectivity observed, along with the control of permeability and efflux, highlight the undeniable value of structural biology and structure-based drug design (SBDD) when the properties of an HTS hit are in a favourable window – even more so when the target candidate profile includes BBB permeability. With a keen focus on SBDD and MTA-cooperative binding conformations, the team at Tango Therapeutics achieved a 3,000-fold increase in potency and 15-fold selectivity over MTAP-null cells, all while adding a mere 83 Da in mass and a LogD reduction of 0.4 units. We eagerly await further news as TNG908 progresses through the clinic.
If you have an interesting drug discovery project you would like to discuss with Domainex’s medicinal chemistry experts, please get in touch.
Reference:
- Discovery of TNG908: A Selective, Brain Penetrant, MTA-Cooperative PRMT5 Inhibitor That Is Synthetically Lethal with MTAP-Deleted Cancers. Kevin M. Cottrell, Kimberly J. Briggs, Douglas A. Whittington, Haris Jahic, Janid A. Ali, Charles B. Davis, Shanzhong Gong, Deepali Gotur, Lina Gu, Patrick McCarren, Matthew R. Tonini, Alice Tsai, Erik W. Wilker, Hongling Yuan, Minjie Zhang, Wenhai Zhang, Alan Huang, and John P. Maxwell. Journal of Medicinal Chemistry 2024 67 (8), 6064-6080