By Sophie Williams
Autoimmune diseases are believed to affect 5% of the population in the western world and are one of the top causes of death for women aged up to 65 years. There are currently over 80 diseases and annual costs for treating seven representative subclasses (Crohn’s disease, ulcerative colitis, systemic lupus erythematosus, multiple sclerosis, rheumatoid arthritis, psoriasis and scleroderma) are estimated to exceed $50 billion in the United States.
Retinoic acid receptor-related Orphan Receptor gamma (RORγ) is a nuclear receptor and is a master regulator of the cell function and development of Th17 (third helper T cells). Th17 is responsible for the production of Interleukin 17 (IL-17), a pro-inflammatory cytokine, which plays a crucial role in chronic inflammation; consequently, RORγ is considered a promising drug target. Currently, there are no RORγ inhibitors on the market despite several examples advancing to human clinical trials over the last decade. Many of the current inhibitors lack sufficient selectivity against other nuclear receptors and exhibit poor physicochemical properties.
Given these risk factors Shiozaki et al.1 employed a two-fold strategy:
- Selecting non-typical nuclear receptor chemotypes for hit optimisation with an emphasis on high three-dimensional (3D) character (Fsp3) to improve nuclear receptor selectivity
- Using drug-likeness indices, such as ligand efficiency (LE) to impart optimal drug-like properties of their inhibitors.
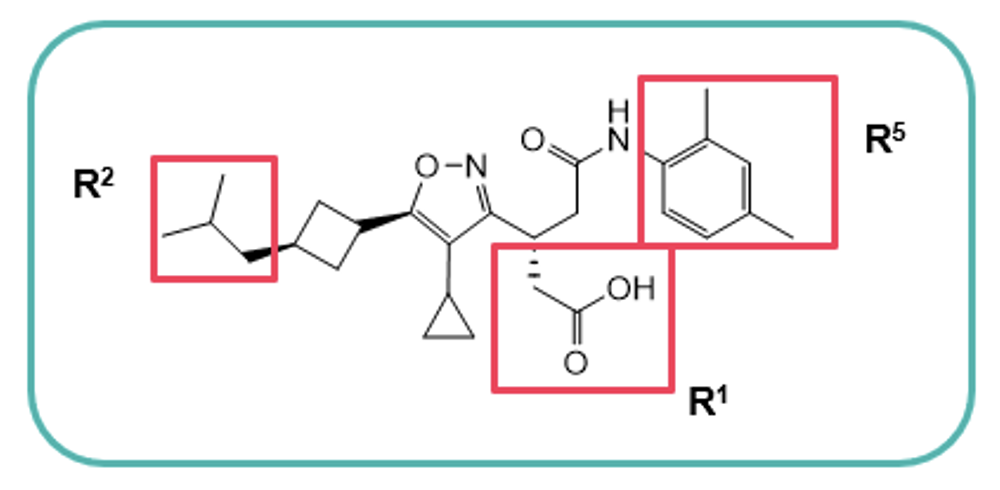
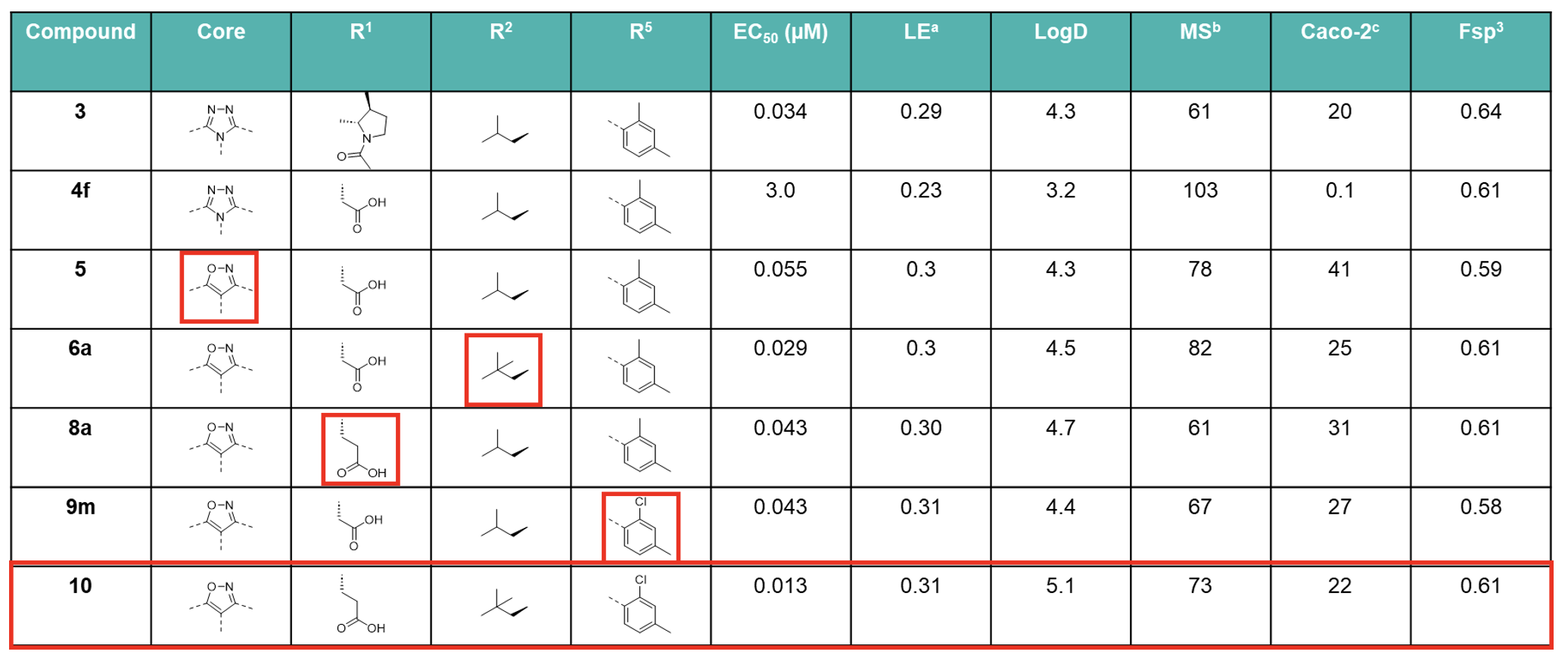
aLE = −1.37 log EC50(LUC hRORγ)/HAC., bhuman liver microsomes (% remaining after 60 min.), cA to B (10−6 cm/s).
The introduction of a terminal neopentyl group at R2 minimised susceptibility to oxidation, thereby increasing metabolic stability (MS), whilst increasing the potency by harnessing extra Van der Waals interactions, maintaining LE and improving cell permeability.
Hydrophilic groups projecting toward the outside of the hydrophobic binding pocket were introduced with the addition of a solubilising acidic group which boosted MS. Core structure scouting identified the best hub module to allocate R2−R5 substituents in the right position and improve the ADME profile. The optimal motifs for R2−R5 with the best balance of LE, MS, and Caco-2 cell permeability were assembled to generate Compound 10, which demonstrated high druglike-character, exhibiting a Fsp3 value of 0.61 and an LE score of 0.31, resulting in on-target specificity and favourable physicochemical properties. Compound 10 was therefore selected as a clinical candidate, JTE-151.
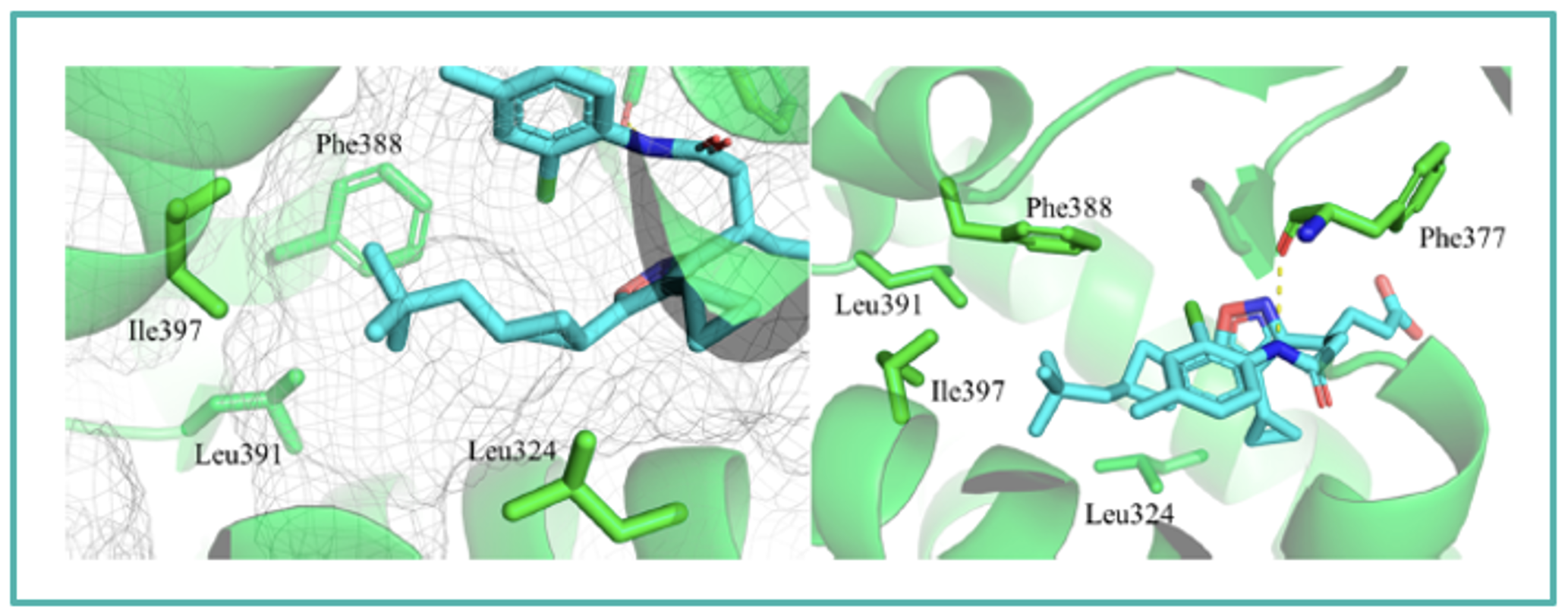
Cocrystal structure of JTE-151 in RORγ (PDB 8X7E)
The binding orientation of JTE-151 in human RORγ was confirmed by X-ray cocrystal analysis, which showed that the compound binds to the ligand binding pocket with a U-shaped conformation. The good RORγ affinity of JTE-151 could be attributed to the terminal amide region, where hydrogen bonding was observed with Phe377 located in the binding pocket of RORγ.
JTE-151 at 3 and 10 mg/kg suppressed IL-17 levels in mice and reduced the number of Th17 cells in the spinal cords, suggesting that JTE-151 suppressed the Th17 cell differentiation. JTE-151 at 10 mg/kg significantly decreased the experimental autoimmune encephalomyelitis (EAE) clinical score (severity of paralysis) and demonstrated better efficacy compared to the anti-IL-17A Ab.
Greater than 100-fold selectivity was demonstrated against antagonist activities at three nuclear receptors (hRORα, hRORβ, hSF1), and both agonist and antagonist activities against a further 12 nuclear receptors (mGR, hAR, hERα, hPR, hVDR, hFXR, mLXRα, hPPARα, hPPARδ, hPPARγ, hRARα, hRXRα). The IC50 values of JTE-151 against a safety screening panel of receptors and enzymes were also found to be > 10 μM. Weak direct inhibition against CYP2C8 (IC50 = 6.8 μM) was exhibited, but the IC50 values for other typical CYPs were >10 μM.
JTE-151 showed a slight hERG liability, with an IC50 of 7.4 μM, however, no QTcF prolongation, a low potential for genotoxicity and one-month GLP-complaint toxicity studies in rats and dogs did not reveal any development-limiting toxicity.
Single dose phase I clinical trial in healthy volunteers at doses of 30 to 1,600 mg demonstrated a dose dependent plasma exposure of JTE-151, with no severe adverse events observed up to the highest dose.
To conclude: JTE-151 is a first in class RORγ inhibitor which has been designed with high 3D functionality for excellent on-target specificity and good LE for “drug-likeness”, and has successfully completed phase I clinical trials.
If you would like to discuss your drug discovery project with Domainex’s medicinal chemistry experts, please get in touch.
Reference:
- Shiozaki et al. Discovery and SAR of JTE-151: A Novel RORγ Inhibitor for clinical development. J Med Chem., 2024, 67, 2, 952-970